

Sharing the Marie Sklodowska-Curie experience
|
In these very unusual times we’re currently living in, the development of a vaccine against SARS-CoV-2 (the coronavirus responsible for the COVID-19 pandemic) has been in everyone’s conversation at some point. We have come to understand how the process of developing and testing a vaccine works and become a bit impatient about the results. Although there are many differences between a vaccine used to prevent an infection, and a drug to treat a condition, I believe it is a good moment to review the various necessary steps it takes to get a drug[1] from being an idea to having it bottled in your nightstand. 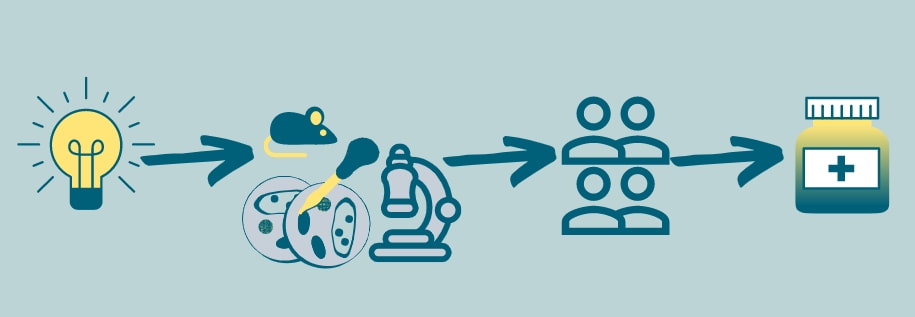 Identification, discovery, development
There are several ways a new therapeutic agent can be discovered. Usually, scientists work to identify a druggable target in a specific medical condition, and then attempt to design and develop a compound that can induce the desired effect on that target. This is a very long and rigorous process that can take several years or even decades, but has the potential of delivering a new treatment, or even a cure for a disease. Other times, the idea to use an agent arises from the observation of its effect on other conditions, as was the case with Sildenafil (now used for erectile dysfunction), and Duloxetine (now used for urinary incontinence), among others. This is referred to as drug repositioning or repurposing. The advantage of this approach is that most of the following steps can be shortened or omitted, as the drug’s safety has already been described for its original use. In this first identification and development stage, only a small proportion of the possible agents are promising enough to go for further research. Preclinical studies After identifying and developing a compound, and before testing it in human patients, preclinical studies are required to assess its safety, efficacy, interactions, and impact in outcomes that are otherwise impossible to determine in humans. This can be done in vitro or in vivo. In the former, the study is carried out in cells or tissues outside of a living organism, which gives us the chance to control the environment, increase throughput, and reduce costs. In the latter, the agent is tested in a living organism, delivering more information about its impact, efficacy, and safety in an actual functioning system. Consequently, it is of upmost importance to develop and characterize animal models that faithfully resemble the human condition and to use species with known similarities to human patients in order to make conclusions from these results easier and anticipate how the agent will act in the clinical scenario. Clinical studies In EU countries the European Medicines Agency (EMA) doesn’t have a role in the authorization of clinical trials. Instead, this is the responsibility of the national competent authorities. In the United States, the Food and Drug Association (FDA) is responsible for authorizing the clinical testing of a new drug. Drug developers must submit an application to the competent authorities including – but not limited to – manufacturing information, chemical and physical properties, how the drug works and how the body processes it, safety and efficacy findings in preclinical studies, and information on the planned clinical trials. After the authorities review the application, a decision is made to stop/delay the investigation or approve a clinical trial. Both the EMA and the FDA offer the researchers assistance in this process. Clinical trials have different phases, with different purposes, duration, and number of participants.
EMA/FDA review After a drug is proven to be safe and effective in controlled clinical trials, the developers submit all the data collected in the previous steps to the EMA or FDA, who then organize a team of reviewers composed by experts in different fields, such as pharmacology, statistics, and other relevant areas. In Europe, the EMA makes a recommendation to the European Commission which then takes a final legally binding decision on whether the medicine can be marketed in the EU. In the US, the FDA review team makes the decision to approve or not to approve the drug. Post-market monitoring Even though the process is long and rigorous, it is impossible to be 100% certain of the efficacy and safety before actually commercializing the drug. This is why monitoring patients that receive the drug for months and years after its already in the marketplace is crucial to monitor possible adverse reactions that may arise. The agencies can request long-term safety studies from drug developers, who are obliged to submit regular reports detailing any adverse events after treatment with the medicine. All these steps have been put in place to ensure we get the safest and most efficient treatments we can, even if it takes several years, or even decades, to achieve. Footnotes [1] Drug, agent, and compound are used as synonyms throughout the text. [2] Side effects are unwanted but documented effects that may arise during the normal use of a drug and are independent of the dose. Adverse reactions are unexpected events that are either unforeseen or dangerous and may or may not be related to the dose. Phase 3 trials are more useful to monitor adverse reactions given the larger number of individuals studied. The author Ignacio Valenzuela is an Early Stage Researcher of iPLACENTA based at Katholieke Universiteit Leuven, in Belgium. Read his earlier blog post here. References “The Drug Development Process”. US Food & Drug Administration. 24 Oct. 2020, https://www.fda.gov/patients/learn-about-drug-and-device-approvals/drug-development-process “From laboratory to patient: the journey of a medicine assessed by EMA”. European Medicines Agency. 24 Oct. 2020, https://www.ema.europa.eu/en/documents/other/laboratory-patient-journey-centrally-authorised-medicine_en.pdf “Development of medicines”. European Federation of Pharmaceutical Industries and Associations. 24 Oct. 2020, https://www.efpia.eu/about-medicines/development-of-medicines/ Leave a Reply. |
About the blogBeing a PhD student in a European training network is a life-changing adventure. Moving to a new country, carrying out a research project, facing scientific (and cultural) challenges, travelling around Europe and beyond… Those 3 years certainly do bring their part of new - sometimes frightening - but always enriching experiences. Categories
All
Archives
December 2021
|
 RSS Feed
RSS Feed

27/10/2020
0 Comments